Omics Analysis with Apache Spark
GTEx eQTL/sQTLデータ編 第1回
今回はApache Sparkを使ったデータづくりを紹介します。Apache Sparkはビッグデータ解析基盤で、多数のサーバーをつなぎあわせ、ひとつの大きなデータ処理を行うことができます。GridEngineやSlurmなどのジョブスケジューラーを使うHPCのシステムとはまた違い、大量のデータを分散処理するために次のような仕組みを基本に動作します。
- データを自動で分割
- 分割されたデータに対するデータ処理が可能
- 上記2つの特徴により、数千台以上でのデータ処理が可能
Sparkで利用可能なソフトウェアとして、バイオインフォマティクスの世界ではHail(http://hail.is) が有名と思いますが、SparkネイティブでVCFなどの変異情報を扱えるGlow(https://glow.readthedocs.io/en/latest/?badge=latest) もあります。また、Beta版の部分はありますが変異の検出で有名なGATKもApache Sparkで動作するように開発が進んでいます。
今回は組み合わせて分析したいデータとして挙げられるGTExのeQTLデータを、Apache SparkのネイティブなDataFrameとして扱えるようにしてみます。
起動
Apache Sparkでは使いたい機能や規模に応じて必要なオプションを設定して起動します。
最初のうちは慣れないかもしれませんが、今回の処理に必要最低限なオプションは以下のとおりです。
| オプション | 役割等 |
|---|---|
| --packages | packages以下にコンマ区切りで依存関係にあるパッケージを指定しています。 |
| io.projectglow:glow-spark3_2.12:1.1.2, io.delta:delta-core_2.12:1.0.1, |
この2行はdeltaを使うために必要です。 |
| nl.basjes.hadoop:splittablegzip:1.3, | gzipされたファイルを並列で扱うために入れています。 |
| org.apache.hadoop:hadoop-aws:3.2.0, | s3へアクセスするために必要なものです。 |
| --conf spark.driver.memory=12G | driverに12GBのメモリを割り当てています。 今回はClusterではないのですがその場合はdriverがexecutorの役割を担うためこのようにしています。 |
| --conf "spark.hadoop.fs.s3a.endpoint=http://[IP Address]:[PORT]" | s3のエンドポイントを指定しています。 今回はオンプレミスなのでminioを使っています。 |
| --conf "spark.hadoop.fs.s3a.access.key=[Key]" | s3のaccess keyを指定しています。 |
| --conf "spark.hadoop.fs.s3a.secret.key=[Key]" | s3のsecret keyを指定しています。 |
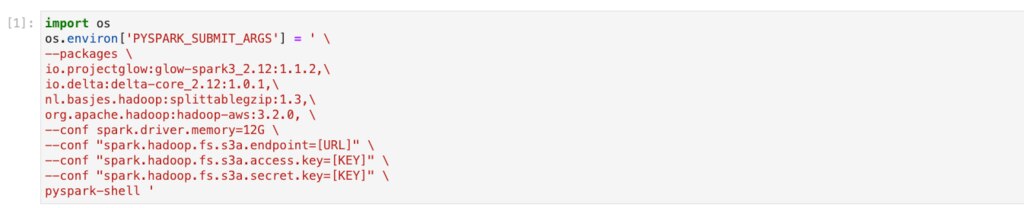
SparkSessionとSparkContextを作成します。
| 命令文 | 役割等 |
|---|---|
| from pyspark import SparkContext, SparkConf, SQLContext from pyspark.sql import SparkSession |
SparkSessionとSparkContextをつくるのに必要です。 |
| sparkConf = SparkConf() sparkConf.setAppName('GTEx eQTL data test') |
Sparkの設定を記述できます。 今回は必要な設定は先のPYSPARK_SUBMIT_ARGSに入れています。 |
| spark = SparkSession.builder.config(conf = sparkConf).getOrCreate() | Spark Sessionを作成しています。 |
| sc = spark._sc | 作成したSparkSessionより、SparkContextを得ています。 |
GTEx eQTLのデータをダウンロード
GTExのeQTLデータ(eQTL Tissue-Specific All SNP Gene Associations https://www.gtexportal.org/home/datasets )はGoogle Cloud Storageでホストされています。
690GBのデータがあり、ダウンロードにはrequester-paysという仕組みを使う必要があります。requester-paysは、「ダウンロードする人が必要な費用を支払う」というものです。 ダウンロードは次のセルにあるように、「-u プロジェクト名」を指定する必要があります。
requester payはダウンロードする度に費用が課金されるので一度実行したらコメントアウトしておきます。 今回はまずAdipose_Subcutaneousのみを取得して、スキーマの検討・作成やDataFrameへの読み込みと試験、ディスクへの書き出し、読み込みの試験を行います。
まだデータも小さいので、Sparkは手元にあったmacOS上で動かしています。
とはいえCloudでも同じコードを使いたいのでminioで構築したs3互換ストレージを使います。

データの確認
4.2GBのgzipされたファイルで、以下のような内容が入っています。
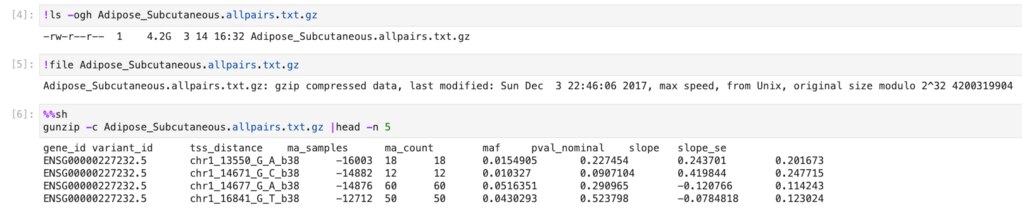
4.2GBのgzipされたファイルで、上記のような内容でした。
データの変換
variant_idは chr1_13550_G_A_b38のように複数の情報がひとつになっていてこのままでは利活用に支障があると思われるため、次のような列に変換をすることにします。
また、後で他のtissueも結合しますので新たに「tissue」を追加します。
| 列名 | 内容例 |
|---|---|
| contigName | chr1 |
| start | 13550 |
| referennceAllele | G |
| alternateAlleles | A |
| tissue | Adipose_Subcutaneous |
スキーマの検討
以下ではスキーマを検討していきます。
SparkのDataFrameではオプションとして便利なInferSchemaがあります。
これはその名のとおりデータからスキーマを類推してくれるものですが、大きなデータに適用するとものすごく時間がかかります。
また、optionにsamplingRatioという便利そうなものもありますが、inferSchemaが有効になっていると効かないので注意が必要です。 (参考: https://spark.apache.org/docs/latest/api/python/reference/api/pyspark.sql.DataFrameReader.csv.html?highlight=inferschema) まず、検討用にデータを小さくしておきます。

以降で使うモジュールをロードし、ファイル名とtissueを定義します。

小さくしておいたファイルを読み込ませます。
ここではinferSchemaを使用し、スキーマを類推させています。
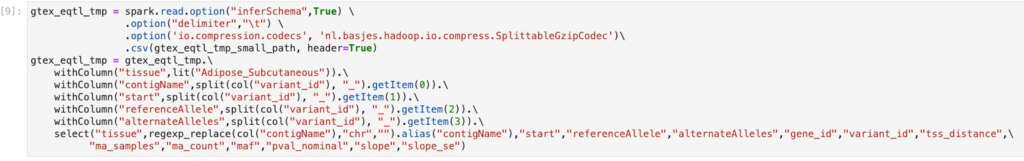
スキーマを確認してみます。
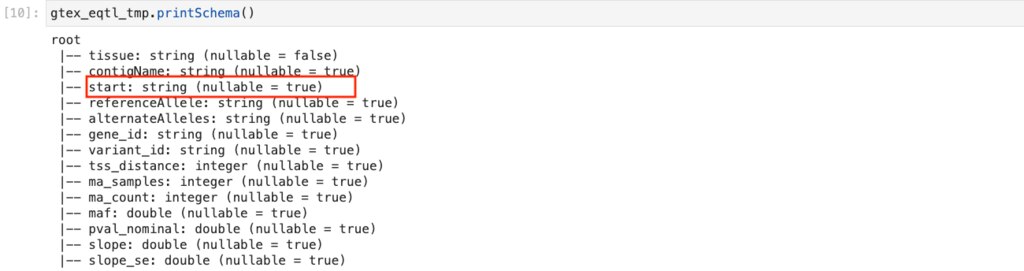
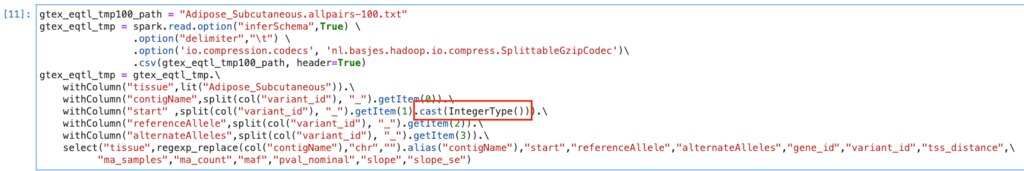
startは整数しかとらないはずである上、範囲を指定したフィルターもしたいデータですので、
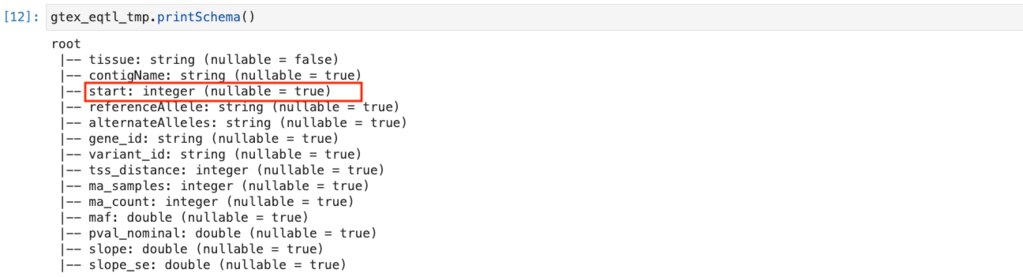
以下のようにIntegerに直します。 .cast(IntegerType()) を付与しています
これでstartがintegerになりました。
スキーマ作成と適用
これを参考に以下のようにスキーマを作ります。

Adipose_Subcutaneousの全データを読み込んでみます。
inferSchemaはFalseにし、schema=schemaで、↑で定義したスキーマを適用しています。
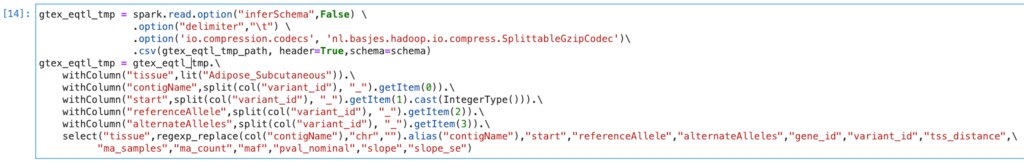
結果を確認します。
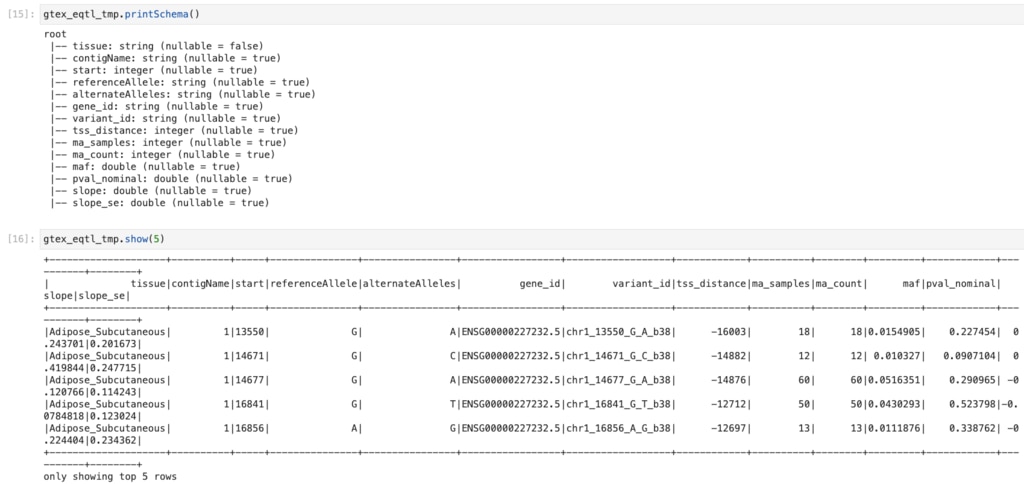
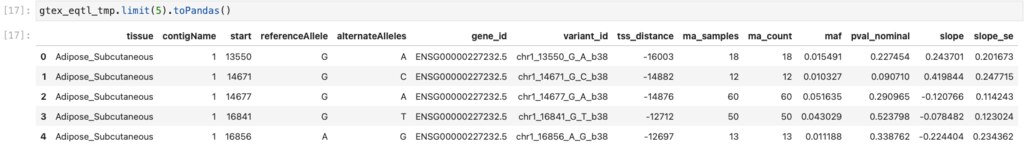
スキーマも変換結果もOKですね。 以下のようにlimitとtoPandasを組み合わせるともっとキレイに表示できます。
ディスクへの書き出し
今回はdelta形式で、s3とmacのローカルディスクの2箇所へ書き出しを行ってみます。
s3への書き出し

ローカルディスクへの書き出し

ディスクからの読み込み
s3

ローカルディスク

DataFrame操作テスト
読み込んだDataFrameは次のようにFilterなどの操作ができます。
ここでは2番染色体のみに絞り込み、pval_nominalをsortしています。
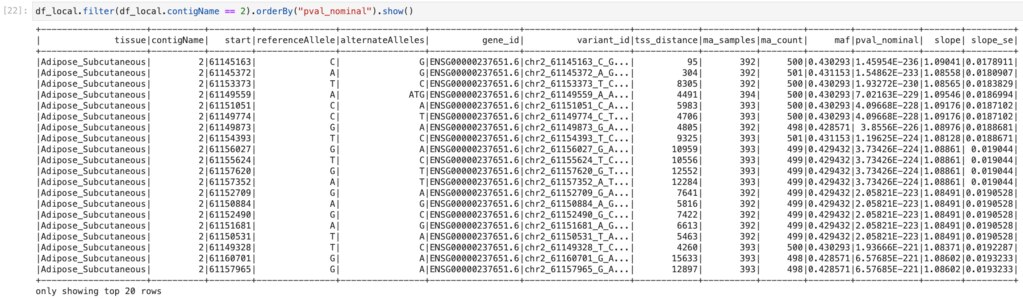
これでGTEx eQTLのAdipose_SubcutaneousをSparkネイティブなDataFrameにすることができました。次回はCloud上にクラスターをつくり、eQTLの全tissueをDataFrameにしてみる予定です。 以上ですがいかがでしたでしょうか?
Apache Sparkでは他にも多様なデータを取り込み解析することができます。
ゲノムデータを中心としたビッグデータ解析基盤にご興味がありましたらぜひお声がけください。